The currently accepted, mainstream theory of cancer
Cancer is taken to be a ‘genetic disease’ at heart. This is because the currently accepted Somatic Mutation Theory (SMT) posits that mutations in the nuclear genome are the fundamental etiological drivers of the disease. These mutation are thought to arise when “an individual mutant clone of cells begins by prospering at the expense of its neighbors” through a microevolutionary process of clonal selection. The nuclear genomes of cancer cells are mutated, disorganized and unstable, and outside their nucleus metabolism is abnormal. Under the SMT umbrella, this metabolic abnormality is assumed to be a downstream symptom of the mutated nuclear genome, a hallmark of the disease but not its causal agent.
A much less popular but competing theory of cancer
A competing theory, the Metabolic Theory (MT) of cancer, effectively reverses this arrow of causality. From nuclear mutations causing abnormal metabolism we got to abnormal metabolism causing nuclear mutations. It postulates that faulty oxidative phosphorylation (OxPhos) – the end stage of cellular respiration – is the root cause of cancer. Consequently, the 6 hallmarks of the disease, angiogenesis, evasion of the immune system, evasion of death, evasion of growth suppressors, invasion and metastasis, and finally sustained proliferative signaling, all follow on from this metabolic abnormality. What the latter presents as is the continued expression of glycolysis in the presence of oxygen, also known as the Warburg effect.
Linking abnormal OxPhos to gene regulatory mechanisms
Whether mutations cause cancer or simply emerge in the cancerous cell as a result of defective OxPhos, it remains important to investigate the links between our genetic profile, both in terms of our base DNA code, gene regulatory networks and epigenetic fingerprints. Initially, genetic features of cancer was limited to abnormal karyotypes, like a different number of chromosomes (or aneuploidy), and somatic mutations. With the advent of epigenetics and a deeper understanding of gene regulatory phenomena, we have at least two additional avenues down which we may potentially learn to better profile cancer cells. This is true regardless of which theory of cancer one operates under.
The question I wish to explore here is Which gene regulatory mechanisms are linked to dysfunctional OxPhos in cancer? The relevant laboratory and software techniques are discussed first, followed by the particular mechanisms underlying the disease, hopefully helping to further elucidate the merits and demerits of both theories.
Definitions: genetic vs epigenetic changes
Before delving into data gathering techniques, a clear and parsimonious exploration of the question necessitates defining genetic, epigenetic and broader gene regulatory phenomena.
Genetic changes
Genetic changes or mutations refer to alterations in base genetic code, whether that be different nucleotides, their order in a sequence, the orientation of a sequence, the translocation of a sequence or the number of times a sequence repeats.
Epigenetic changes
As for epigenetics, in 1952 Waddington used the term quite broadly “to refer in general to what we now know to be changing patterns of gene expression that underlie development and that are oſten triggered by signals sent from other cells”. According to the 2014 6th edition of Molecular Biology of the Cell, the covalent modification of histones in nucleosomes falls under an epigenetic “form of inheritance that is superimposed on the genetic inheritance based on DNA”. Mark Ptashne argues that the covalent modifications of histones is not an epigenetic mechanism given that “the enzymes that impose such modifications lack requisite specificity; the modified states are not self-perpetuating; and the roles played by the modifications remain for the most part obscure”. Experimentation tells us that “histone modifications are not maintained as cells divide”. These histone modifying enzymes are part of a response to epigenetic regulation and not a cause thereof. According Ptashne’s criteria, histone modifications fall under the more general category of gene regulation, leading to a more narrow definition of epigenetics. This definition includes the phenomena of memory (“continual activities of the specific regulators to maintain that state of expression”) and specificity (“activating one gene or set of genes and not another”). One epigenetic mechanism may employ a positive feedback loop (such as the auto-activation of the cl gene) and another, a negative feedback loop (like the auto-repression of the cl gene). Examples of epigenetic in nature include viral immunity passed down to the next generation or transient inflammatory stimuli causing chronic inflammation via feedback-loops mediated by sequence-specific microRNAs (or other regulatory proteins).
Gene regulatory changes
Gene regulatory changes result from the activity of gene regulatory networks governing the gene expression levels as seen in mRNA and proteins. These networks respond to environmental cues that could be anything from food and light to psychosocial events and drugs.
Detecting cancer
Now that we’ve got our definitions lets talk about about techniques to detect cancer. An early cancer diagnosis, or better yet anticipating the transition of a benign tumor to a malignant one, is of utmost importance in reducing cancer associated deaths. For this reason, assays detecting DNA methylation patterns regulating gene expression and activities of their upstream enzymatic effectors have become of great interest. They are the subject of Shinjo and Kondo’s 2015 paper upon which most of the following analysis is based. A point of note, unlike Mark Ptashne these authors consider DNA methylation to be an epigenetic phenomenon.
DNA methylation
DNA methylation pattern assays first require differentiating methylated DNA from unmethylated DNA, which can be done physically, using a methyl-binding column, or by inducing sequence changes through bisulfite treatment, for instance. Other techniques based on sequence changes include the use of restriction enzymes of varying sensitivity to DNA methylation sites, as well as methylated DNA enrichment with anti-cytosine or methyl-binding anti-bodies. DNA methylation appears to be stable in cancers and can thus be relied upon to establish methylation patters related to gene expression.
DNA methylation pattern assays can be conducted on samples of cell-free DNA (cfDNA) found in blood, stool, urine, tissue and other bodily fluids. Although cancer cells resist apoptosis, the disease process results in higher overall rates of apoptosis and tissue necrosis. This leads to increased levels of cfDNA circulating in the blood which can be indicative of a loss of heterozygosity and the presence of mutations. For example, blood samples of cfDNA have shown RASSF1A DNA demethylation associated with increased sensitivity to cancer treatment. The converse is also true, decreased sensitivity to treatment is associated with increased methylation.
Methylation tests on fecal, urine and sputum samples each use different biomarkers and do not all perform equally well. For stool samples, Cologuard™ uses NDRG4, BMP3 and KRAS mutation methylation biomarkers showing 92.3% sensitivity and 86.6% specificity. For urine samples, the GSTP1 methylation biomarker shows 88% sensitivity and 60% specificity. For sputum samples, the Epi prolung test uses the SHOX2 methylation biomarker showing 60% sensitivity and 90% specificity.
Gene regulatory imprints
Cancer treatments directly targeting gene regulatory mechanisms broadly cover 3 areas; histone modifications, DNA methylation associated pathways and readers of gene regulation.
Regarding the first area, there are 18 known histone deacetylase inhibitors (HDIs) divided into classes 1 through 4 with which to work with. They stop the deacetylation of lysine (K) and are associated with the restoration of silenced genes, the induction of growth arrest, the interruption of differentiation and the prompting of tumor cell apoptosis. HDI based cancer treatments are still quite new. Currently there are only 3 FDA approved HDIs in the United States of America: Vorinostate, Romidespine and Belinostat.
Regarding the second area, DNA methyltransferase inhibitors (DNMT1 inhibitors) are equally novel and only one has been approved for myelodysplastic syndromes (MDS) therapy – bone marrow cancers. The mechanism of action of another such molecule is illustrated in Figure 1 below, taken from Shinjo and Kondo’s paper. IDH1 usually catalyzes the oxidatively decarboxylation of isocitrate to α-ketoglutarate (α-KG) which has for effect of replenishing the TCA cycle. IDH-1 mutants divert α-KG towards the catalysis of oncometabolite 2-HG. As the latter accumulates it inhibits α-KG-dependent dioxygenases, histone demethylases and TET family proteins. IDH-1 inhibitors therefore interfere with 2-HG accumulation.
The third area is concerned with cancer treatments exploiting molecules capable of interfering with gene regulatory ‘reading’. Bromo and Extra Terminal proteins (BETs) read the code of histone modification, particularly acetylated histones. BET inhibitors bind particular residues of histone modification, effectively out-competing bromodomain-competing proteins (BRDs) of the BET family. For example, fusion oncoprotein BRD4 is displaced from chromatin by BET inhibitor I-BET151, thereby decreasing the oncogenic transcription potential of BCL2, C-MYC (cytoplasmic myelocytomatosis oncogene) and CDK6.
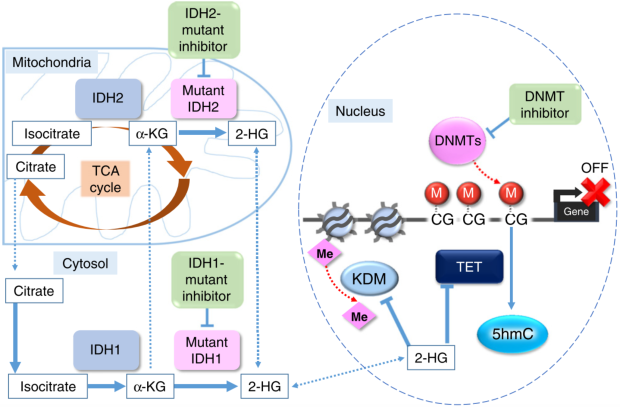
Figure 1. IDH1-mutant
Chromatin Immunoprecipitation Sequencing (ChIP-seq) assays are used to map the genome-wide binding of gene regulatory proteins. For example, this technique reveals the different binding patterns of the Sirtuin1 protein (Silent Information Regulator 1) in the nucleus accumbens of rats before and after the administration of cocaine over 7 days. ChIP-seq in conjunction with RNA and immunoblotting assays can link posttranslational modifications made to newly synthesized histones and the subsequent patterns of gene expression found at specific genomic loci affected by this chromatin assembly. Yang et al. applied this combination of techniques to show ”how one modification that occurs on newly synthesized histone H3, acetylation of K56, influences gene expression at epigenetically regulated loci in Saccharomyces cerevisiae”. Essential information about cellular behavior is obtained with ChIP-seq. Other programs are then used to map relationships between these patterns of immuno precipitated chromatin.For example, RNA-seq visualization tool Cascade does just that for copy number variants (CNVs) and the MAP2K4 pathway of ovarian cancers, visible in Figure 2 below. It can then form the basis of further experimentation involving the in vivo alteration of chromatin patterns associated with malignancy, in the hopes of impeding tumor progression.

Figure 2. RNA-seq visualization tool Cascade for ovarian cancer CNVs & MAP2K4 pathway
Sirtuins
The first mechanism explored here involves sirtuin proteins 1 to 7. According to Mark Ptashne’s their criteria fall under the category of gene regulation. Amongst the 7 sirtuin proteins, those of particular note are sirtuins 3, 4, 6 (and 7 to a lesser degree) because of their pronounced ability to suppress the Warburg effect as a function of their gene regulatory capacities. Sirtuins are NAD+-dependent class III histone deacetylases (HDACs) catalyzing ADP-ribosylation reactions which can loosen chromatin. This effectively makes DNA more or less accessible, as well as facilitate or hinder the activities of histone effector proteins. These sirtuins are most directly involved in cancer via their action on the Warburg effect. This is because their activities depend on the NAD+/NADH ratio, a general indicator of metabolism, and more specifically cellular redox state and energy status. NAD (the reduced form of NAD+) and NADH are physiological sirtuin inhibitors whilst NAD+ and resveratrol are sirtuin-activating compounds (STACs). The following discussion of sirtuins 1 to 7 is based on Kleszcz et al.’s 2015 paper as well as Douglas Wallace’s 2014 NIH presentation “A Mitochondrial Etiology of Metabolic and Degenerative Diseases, Cancer and Aging”.
Sirt6
SIRT6 is found only in the nucleus and is particularly interesting amongst sirtuins, in large part because of its co-repression of transcription factor MYC (myelocytomatosis oncogene). This repression is associated with both the inhibitions of glycolysis and ribosomal activity. In cancer cells, the nucleoli containing ribosomes have the striking morphological feature of being engorged due to increased biosynthetic demand. Non-transformed cells lacking SIRT6 become tumors when glycolysis is intact but fail to do so when it is abolished, a pattern which strongly argues that the Warburg effect drives tumorigenesis. Like for most sirtuins, SIRT6’s tumor-suppressing effect depends on the cell type and disease stage. Nevertheless, SIRT6 over-expression affects chromatin structure such that cancer cells will apoptose but normal cells won’t. One way this is thought to occur is through the H3K9 deacetylation of the Hypoxia-Inducible Factor-1α (HIF-1α) gene promoter. When HIF-1α is stable in cancer cells pseudorespiration is ongoing. This occurs when ATP is produced through mitochondrial fermentation involving substrate-level phosphorylation. The cancer cell is less sensitive to apoptotic signals when the HIF-1α complex is stabilized. Fortunately, SIRT6 over-expression can destabilize it. Furthermore, mouse models of liver cancer have shown that the ‘genome guardian’ tumor suppressor p53 can induce SIRT6 expression so as to inhibit hepatic gluconeogenesis. This underscores a mechanism for p53 in the maintenance of glucose homeostasis.
Sirt3
Other sirtuins affect HIF-1α stability such as SIRT3 which is found both within the mitochondrial matrix and nucleus under normal growth conditions. Downregulation of its activity effectively reprograms metabolism such that an increased level of reactive oxygen species (ROSs) signals HIF-1α stabilization. This in turn upregulates glycolytic enzymes. SIRT3 impedes aerobic glycolysis and thus the production of lactate through an additional pathway involving pyruvate dehydrogenase E1α (PDHA1). The deacetylation of lysine 321 on PDHA1 will increase its activity, diverting and increasing glucose utilization through the Citric Acid Cycle (TCA). Like SIRT6, SIRT3 appears to suppress or promote tumorigenesis depending on the cell type as well as the confluence of stress and apoptotic stimuli. Inflammation is one such stressor typically present in tumors and can be attenuated by fasting in human subjects. SIRT3 is NAD-dependent (or ‘nutrient sensing’) and when fasting, its activity diminishes. This has for consequence of blunting the response of the NLRP3 (NOD-Like Receptor family Protein 3) inflammasome.
Sirt4
Metabolism is inextricably interwoven with biosynthetic demand given their functional interdependency. This is exemplified in the interaction of the mammalian target of rapamycin complex 1 (mTORC1) and SIRT4. The latter is only located in the mitochondrial matrix and is involved in balancing the oxidation and synthesis of fatty acids. mTORC1 however is a general growth promoter. Its activation will upregulate glutamate dehydrogenase (GDH) via SIRT4 repression and in so doing enable cancer progression. This enhanced glutamine metabolism both replenishes TCA cycle intermediates required for biosynthesis as well as serve as fermentable substrate contributing to filling the energy gap resulting from the cancer cell’s defective oxidative phosphorylation. Interestingly, sufficiently extensive DNA damage can induce SIRT4 expression and shut down glutamine metabolism, a requisite process for cellular repair. This sirtuin appears to suppress the formation of tumors by inhibiting excessive glutamine metabolism and maintaining genomic stability.
Sirt1
SIRT1 is the most highly conserved sirtuin in mammals and is found both in the nucleus and cytoplasm. It modulates the metabolism of lipids and glucose in the liver, insulin secretion in the pancreas and engenders fat mobilization in adipose tissue. All of these features together point to SIRT1 as a key metabolic sensor. SIRT1 negatively regulates phosphoglycerate mutase 1 (PGAM1) which is upregulated in many cancers and occupies an important point in the glycolytic pathway where many glycolytic intermediates upstream of it serve as biosynthetic precursors. SIRT1-mediated deacetylation of PGAM1 will thus reduce glycolysis, flux down the pentose phosphate pathway (PPP). The levels of the aforementioned biosynthetic intermediates will thus be lower too. This all serves to inhibit tumor growth given the tumor’s reliance on these factors. Like other sirtuins, SIRT1 also demonstrates tumor suppressive action mediated by HIF-1α deacetylation. This sirtuin, like others in its family, has its tumor suppressing capacities contrasted by context-dependent tumor promoting ones. Through its deacetylating activities it can promote tumorigenesis by inhibiting p53, p73 and HIC1 (Hypermethylated In Cancer 1). Given how common it is for genes and gene regulating molecules to both suppress and promote tumorigenesis, they should not be viewed as solely doing one or the other, but rather as sitting at the intersections in cell pathways leading towards or away from growth. The same analogy can be applied to the gene themselves, implying that terming them ‘oncogenic’ may have been premature and now thoroughly confusing.
Sirt2
Along with SIRT1, SIRT2 is also located in both the cytoplasm and mitochondria. It is yet another sirtuin that, when overexpressed, can destabilize HIF-1α under hypoxic conditions. Experiments with cells that do not express SIRT2 corroborate this fact given that they fail to affect HIF-1α levels. SIRT2 positively regulates PGAM as opposed to SIRT1’s negative regulation. SIRT2 deacetylates K100 (lysine100) on PGAM, thereby upregulating its activity and thus glycolysis. This sirtuin is mainly found in the cytoplasm and shuttles back and forth to the nucleus. The significance of sirtuin localization is not yet known. However it is interesting to consider the possibility that the direction in which they carry out their activities (either from nucleus to cytoplasm or cytoplasm to nucleus) might explain their context-dependent pro- or anti-tumorigenic effects. During the study of breast tumors, SIRT2 was shown to have both tumor suppressive and promoting capabilities. Here however, tumor grade appeared decisive rather than cell type.
Sirt5 and sirt7
Both SIRT5 and SIRT7 are lesser studied sirtuins. The former is only found in mitochondria where it acts as a global regulator of succinylation. Through its dessucinylation of superoxide dismutase (SOD1) it can lower the level of ROS produced by cancer cells. These ROS are thought to result form increased glycolysis, mitochondrial fermentation in defective mitochondria with poor oxidative phosphorylation. Found only in the nucleus, lysine deacetylase SIRT7 targets H3K18. The latter is found in the vicinity of multiple gene promoters involved in tumor suppression. Similar to the SIRT6, it attenuates engorged nucleoli containing ribosomes in a MYC-dependent manner by deacetylating MYC transcript targets of ribosomal proteins. SIRT7’s maintenance of H3K18 acetylation patterns appears important in keeping this morphological cancer phenotype. Enhanced rRNA production is such a conspicuous feature of cancer that it has been proposed as the 7th hallmark.
Returning to the idea of sirtuin localization being able to potentially explain opposing actions of sirtuins, SIRT7 is present and highly active in the ribosome-containing engorged nucleoli. This suggests its activity may promote rRNA synthesis and thus ribosome biogenesis. Finding a compound capable of inhibiting SIRT7 compound is of great interest. Nevertheless, STACS may be generally preferable to to sirtuin inhibitors for 3 reasons. Activators need not be as potent as inhibitors due to downstream signal amplification, they are more selective due to their ability to bind non-catalytic sites of target proteins, and their mimicry of natural activators is liable to induce fewer side-effects. Unfortunately, Kleszcz et al. make clear that until now (2015) “it has been a conundrum that such a diverse set of small molecules can activate SIRT1, but so far no activators of SIRT2-7 have been described”.
The sooner we learn to exploit endogenous sirtuin activation, improvements in cancer prevention as wellaas palliative and adjunctive cancer care may be attainable. Much of this may stem from appreciating how dietary choices and sleep patterns, for instance, affect the expression of sirtuins. Diet is always front and center as a lifestyle factor and interestingly, multiple long-chain fatty acids have been shown to induce a 35-fold increase in SIRT6 activity at physiological concentrations. Furthermore, mice given nicotinamide riboside benefitted from increased insulin sensitivity, an effect that may stem from nicotinamide riboside’s ability to increase NAD+ levels, an endogenous sirtuin activator. This effect correlated with SIRT1 and SIRT3 upregulation.
Talking about ‘oncogenic activity’ makes more sense than talking about ‘oncogenes’
When cancer is viewed through the genetic paradigm, a parallel between the inconsistent tumorigenic effects of sirtuins and those of oncogenes emerges. A particular sirtuin might flip from being anti-tumorigenic to tumorigenic depending on the tissue type or grade of cancer. An oncogene can be both pro-tumorigenic and suppress tumors without a satisfying explanation as to why. Supporting this latter point is Soto and Sonneschein’s observation that “a mutation that should have produced uncontrolled cell proliferation resulted in cell death or arrest of cell proliferation”. The significance of this parallel is that the opposing actions of both (onco)genes and sirtuins in the genesis and progression of cancer highlights cracks in the framework of SMT. For instance, genetic analyses of BRCA variants only explain about 10% of breast cancer susceptibility and amount to our best current predictions as of 2016. Fortunately, BRCA1’s known interaction with enzyme acetyl coenzyme A carboxylase alpha (ACCA) provides a clearer path for exploiting a metabolic vulnerability than purely genetic analyses of BRCA1 do.
The oncogenic paradox
SMT suffers from poor predictive power in terms of cancer occurrence and treatment outcomes. This led some to wonder how such a specific process (cancer) can stem from a number of unspecific mutation-inducing events (radiation, viruses, inflammation etc.). Albert Szent-Györgi termed the latter phenomenon the “oncogenic paradox”. Fortunately, this paradox and oncogenic inconsistency can be stress-tested by exploring a prediction of SMT:
transferring transformed (cancerous) nuclei into non-transformed (normal) cytoplasm will induce tumorigenesis in the latter
Such nuclear-cytoplasmic transfer experiments have been carried out. They show that nuclei from cancerous cells transferred into normal ones do not reliably transform the cell. The converse of that experiment should yield similar results, whereby cells with tumor nuclei should revert back to a normal non-cancerous state when cytoplasmic contents of normal cells, specifically their respiration competent mitochondria, are transferred to them. In 1988 Israel and Schaeffer showed exactly that. He used cytoplasmic hybrid, also known as cybrids, from preparations of rat liver epithelial cells. Both kind of transfer experiments are depicted in Figure 3 below.

Figure 3. Nuclear/cytoplasm transfer experiments
Kulesa et al. demonstrated “the ability of adult human metastatic melanoma cells to respond to chick embryonic environmental cues, a subset of which may undergo a reprogramming of their metastatic phenotype”. Thomas Seyfried of Boston College summarizes results from multiple nuclear-cytoplasmic experiments, saying “nuclei from cancer cells can be reprogrammed to form normal tissues when transplanted into normal cytoplasm despite the continued presence of the tumor-associated genomic defects in the cells of the derived tissues”. This suggests that something in the cytoplasm, rather than something in the nucleus is driving tumorigenesis. It is this in addition to Warburg’s nearly century old observations of cancer cells displaying some degree of defective OxPhos, that begs the following question
how do gene regulatory elements link defective mitochondria and impaired respiration to highly unstable, disorganized and mutated nuclear genome?
Impaired respiration
ROS
One manifestation of impaired respiration is excessive reactive oxygen species (ROSs). The electron transport chain (ETC) in the inner mitochondrial membrane (IMM) uses electron carrying molecules to synthesize ATP and some of these electrons also (fully) reduce water. This imperfect process also partially reduces water, whereby electrons become unpaired in oxygen’s outer orbital and form multiple ROS compounds, such superoxide anions, hydrogen peroxides and hydroxyl radicals. Hydroxides (OH–) for example may ‘steal’ electrons from membrane lipids and thus initiate a chain-reaction of neighboring lipids stealing electrons from one another. This changes the properties of membranes in an uncontrolled manner. ROS are physiologically unavoidable and participate in cell signaling. Yet because they are inherently damaging to cellular components they must be managed to avoid disease and accelerated ageing. Additional manifestations of impaired respiration include abnormalities in mtDNA, the proton motive gradient (ΔΨm) and the TCA cycle.
The Retrograde Response
Yeast and animal cells have ‘surveillance systems’ to react to these signs of impaired respiration. One of these signs is called the retrograde (RTG) response. The RTP “responds to mitochondrial dysfunction by adapting cell metabolism to the loss of tricarboxylic acid (TCA) cycle activity”. Thomas Seyfried correctly categorizes the RTG response as an epigenetic mechanism because it fulfills the criteria of specificity and memory: the basic helix-loop-helix-leucine zippers Rtg1/Rtg2 transcriptional factors complex demonstrate specificity when binding to the DNA R Box binding site for the transcription of a particular set of genes, as well as memory given that yeast maintains those changes during cellular division when extending their lifespan. The RTG response is a signaling pathway relaying information about the organelle’s status to the nucleus. It’s response determines whether or not the major cell-quality control mechanisms of mitophagy and autophagy are induced.
The RTG response is a central mechanism underlying the MT of cancer because it links the hypothesized metabolic origin to the characteristic abnormally unstable and mutated nuclear genome. Essentially, the RTG response upregulates non-oxidative metabolic networks (or oncogenes from the gene-centric perspective) which shifts the cell’s energy yielding pathways to a predominant mix of glycolysis and glutamine fermentation so as to maintain ATP homeostasis (stable ΔG’ATP) despite insufficient respiration. Figure 4 below is Thomas Seyfried’s illustration (adapted from Michal Jazwinski’s work in yeast) of how the mammalian RTG response occurs.

Figure 4. RTG Response
Normally respiring cells have a dormant RTG response located in the cytoplasm. The RTG is actually a complex composed of Rtg1 dimerized to a strongly phosphorylated Rtg3. When a cell experiences defective or insufficient respiration, cytoplasmic Rtg2 partially dephosphorylates the Rtg1/Rtg3 complex, prompting both proteins to enter the nucleus so that Rtg3 may bind the R Box, followed by Rtg1 re-engaging Rtg3, effectively awakening the RTG response. This leads to the targeted transcription of multiple metabolic and antiapoptotic genes including CHOP, MYC, TOR, NF-𝜅B, Ras and CREB. This non-exhaustive set of genes has multiple associations with cancer initiation, maintenance and metastasis. For one, MYC has been associated with increased levels of ROS and the inhibition of tumor suppressor p53. An example pertaining to metastatic behavior links the RTG response to increased levels of Matrix Metalloproteinase 2 (MMP2), as seen in activated macrophages hybridizing to neoplastic epithelia.
It is important to note that RTG response mediated destabilization of the nuclear genome, as well as the induction of aneuploidy and somatic mutations, all strong favor the MT rather than SMT theory of cancer. Furthermore, the RTG response is also associated with multi-drug cancer resistant phenotypes, increased cytoplasmic calcium, increased ROS, iron-sulfur complex abnormalities, decreased mitochondrial ATP production as well as a lowered ΔΨm. Although there is no direct equivalent in higher eukaryotes of the yeast Rtg2 mitochondrial sensor transducing mitochondrial signals, authors Srinivasan et al. explain that “strong homologies between inhibitors and pathways of both [RTG genes and NF-κB] leads one to believe that the retrograde response is a potential predecessor of the now-central stress-regulator, NF-κB”. In that same paper by Srinivasan et al., reproduced here in Figure 5, one can see the nearly identical RTG and NF-κB activity levels as they relate to 6 major metrics of cellular behavior.

Figure 5. RTG vs NF-kB
Jazwinski also comments on the many similarities between yeast and human RTG responses, adding that “mammalian cells not only display many of the molecular features of yeast retrograde signaling, but they also present the cellular outcome of extended life span that characterizes the retrograde response”. Lifespan extension mechanisms are inextricably tied to tumorigenic control ones. In figure 6 below Klement and Champ illustrate the mechanistic overlap between ketogenic diets (KDs), which can suppress tumors, and calorie restriction (CR), which has been shown to extend lifespan in multiple animal models.

Figure 6. CR & a KD target the same molecular pathways
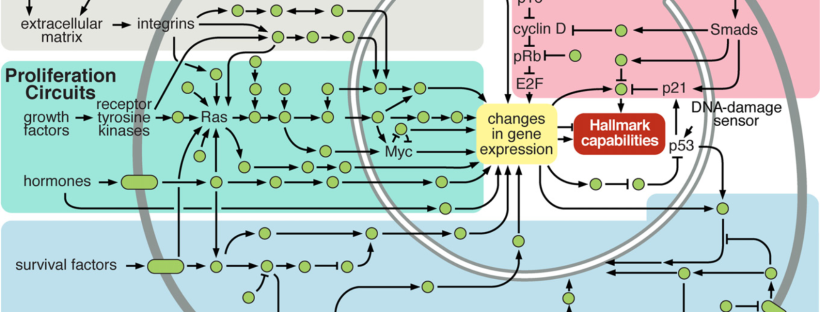
Figure 7 below illustrates the integrated circuits of a cell. What is interesting is that cellular circuitry is obviously drawn through the lens of the SMT. Pathways (arrows) leaving mitochondria towards ‘DNA Damage Sensors’ are nowhere to be seen. There seems to be little acknowledgement of how defective OxPhos leads to direct or indirect upregulation of glycolytic and fermentative networks. Rather, this shift in energy metabolism is deemed to originate from nuclear mutations.
Re-imagining Figure 7 through the MT lens, the RTG response would constitute a ‘Mitochondrial Damage Sensor’ with an arrow leading to NF-κB and integrating back into the web of arrows already present. Furthermore, there would be an increase in the proportion of arrows going from the mitochondria to the nuclear genome (and back), highlighting the metabolic origin as well as the bi-directional cross-talk enabling nuclear genomes to upregulate aforementioned metabolic networks. This is not a baseless hypothetical. For instance, the classic view of p53 as solely regulating tumorigenesis (and thus genomic stability) by inducing mitochondrial apoptosis or via transcriptional factor response elements must be updated, because it is now known to also regulate transcriptional target SCO2 (Synthesis of Cytochrome c Oxidase 2) through which mitochondrial energy production is influenced. Genomic stability, by way of p53, is thus highly dependent upon OxPhos status. Integrating this into Figure 7 once again, this would look like an arrow leaving the mitochondria to SCO2 and then on to p53.

Figure 7. Cellular Circuitry (as of 2000)
Returning to Jazwinski’s paper on the RTG response, another gene regulatory (not epigenetic) mechanism lending support to MT of cancer can be inferred from exploring how ceramide synthase activity found in the endoplasmic reticulum (ER) relates to mitochondrial dysfunction. The hydrolysis of sphingomyelin by sphingomyelinase (Smase) is a catalyzing reaction generating ceramide, a structural and signaling cellular component of mammalian cells. Ceramide appears to be stimulated by a variety of stressors such as TNF-α/matrix metalloproteinases/ROSs, cannabinoids and ionizing radiation. Ceramide is linked to retrograde signaling by way of it prompting Isc1 (Inositol phosphosphingolipase C), leading to autophagy. Jazwinski explains that “sphingosine and ceramide are precursors of complex sphingolipids [which] suggests that the balance in sphingolipid biosynthetic activity can tip the scale in autophagy from quality control to wholesale degradation and remodeling”.
Cannabinoids
In fact, phytocannabinoid THC ((−)-trans-Δ9-tetrahydrocannabinol) was shown to induce apoptosis in vitro. Authors explain this, saying the “first step in the apoptotic pathway was ceramide production, and that this led to loss of membrane potential, and caspase activation, respectively”. Lesser known phytocannabinoid CBD (cannabidiol) was shown by McAllister et al. to reduce the aggressiveness of breast cancer cell proliferation, invasion, and metastasis by affecting mitochondrial ROS production in vitro. This CBD-mediated effect appears to occur down 2 pathways: ERK (extracellular signal-regulated kinase) activation and increasing ROSs further. Both of these pathways can decrease Id-1 (Inhibitor of DNA Binding 1) gene expression, leading to reduced cell proliferation and invasion. The way in which these cannabinoids seem to work adds to the evidence supporting a theory of cancer based in metabolism. One way these cannabinoids exert effects on metabolism involves ceramide, as “ceramide generated upon CB1 cannabinoid receptor activation may enhance ketone body production by [rat] astrocytes independently of MAPK”.
Cannabinoids to ketone bodies
Interestingly, this line of investigation leads to promising in vitro and in vivo experiments highlighting the emerging role of ketone bodies as alluring metabolic fuels. They appear capable of simultaneously stressing cancer cells and supporting normal ones. The link between the MT of cancer and gene regulatory effects of ketones are apparent when considering some of the properties of endogenously produced ones, such as d-β-hydroxybutyrate (βOHB) (chemically not a ketone but a carboxylic acid). βOHB specifically inhibits class I HDACs. This βOHB-induced HDAC action “correlated with global changes in transcription, including that of the genes encoding oxidative stress resistance factors FOXO3A and MT2 […] consistent with increased FOXO3A and MT2 activity, treatment of mice with βOHB conferred substantial protection against oxidative stress”. The shared mechanisms via which both cannabinoids and ketones place metabolic stress on cancer cells whilst protecting normal ones strongly support a MT of cancer.
Conclusion
In conclusion, it is worth pausing for a moment and reflecting on the fact that ketones and cannabinoids, 2 highly non-toxic compounds endogenous to humans, have been unfairly maligned. The former because of its conflation with diabetic ketoacidosis and the latter because of harms associated with drug abuse. Yet, both are now resurfacing in medical research as highly promising compounds, especially for treating the emperor of all maladies, cancer.
It is also worth pondering how the range of new cancer therapies like HBOT (hyperbaric oxygen therapy), ketogenic diets and deuterium (2H) depleted water (DDW) share overlapping mechanisms directly impacting metabolism by influencing mitochondrial status – especially OxPhos activity. The latter therapy, DDW, delayed prostate cancer progression in a 4-month phase 2 clinical trial that was double-blind and randomized. The mechanism of action of DDW as explained by Boros et al. de facto serves as further evidence of a metabolic origin of cancer:
“The excessive appearance, i.e. accumulation of ‘‘metabolically dry” oncometabolites [such as 2-HG] is consistent with our hypothesis that cancer is formed on the basis of mitochondrial defects that lack hydration of TCA cycle intermediates with low deuterium matrix water as the result of such defects. Such claim is supported by the fact that restoring hydratase function of mitochondria reverses tumor cells back to their genetically stable non-proliferating normal phenotype [37] with normal matrix water content, composition and morphology”
It is time for SMT-centric mainstream cancer researchers to reconsider the fundamental mechanisms of action of the few marginally successful gene-based therapies on the market. Rather than viewing genetic, epigenetic and gene regulatory phenomena as epicentres of action of ‘gene-based therapies’, these might actually be downstream of metabolic disturbances. This is a major distinction in how one thinks cancer is caused.